718
Views & Citations10
Likes & Shares
Mucopolysaccharidosis type VI, is a rare genetic
disease caused by a deficiency of the N-acetylgalactosamine-4-sulfatase enzyme,
also known as aryl sulfatase B. In Colombia, there are 36 known cases, of which
10 are in an indigenous group with an estimated prevalence of 1/1,140,000. New
mutations are published continuously and, to date, 140 have been reported. The
main objective of this study is to characterize, by molecular genetics, two
patients identified in Southwestern Colombia (Department of Cauca) with the
severe form of MPS type VI. A single nucleotide (p.C447F) pathogenic
transversion producing a sense change mutation was found in the two index cases
on exon 8 of the ARSB gene. This
gives rise to the exchange of one amino acid for another on the minor domain of
the enzyme: position 206,029 (T/T) TGT>TTT. An unusual frequency of genetic
diseases is found in the department of Cauca in Colombia. In this study, the
two index patients exhibit the same mutation, suggesting the possibility of a
common ancestral allele, probably due to the relative inbreeding and the
geographical isolation of these regions. The above highlights the importance of
public health policies in our country, genetic counseling, neonatal screening
and identification of new cases in areas where incidence is above average.
Keywords: Lysosomal
diseases, Muco polysaccharidosis type VI, Matoreaux-Lamy syndrome
INTRODUCTION
Mucopolysaccharidosis type VI (MPS VI) or
Maroteaux–Lamy syndrome (OMIM #253200), is a rare genetic disease caused by a
deficiency of the N-acetylgalactosamine-4-sulfatase enzyme, also known as
arylsulfatase B (ARSB) (coded on locus 5q11-13) [1]. This enzyme is involved in
dermatan glycosaminoglycan and chondroitin sulfates catabolism [2]. Incidence
can vary among different populations and geographical regions, ranging from 1
in 238,000 births and 1 in 1,298,000 births [3]. In Colombia, of 36 known
cases, 10 are in an indigenous group (Author data, not published yet). This
equates to an estimated prevalence of around 0.08/100,000 inhabitants for the
general population of Colombia; 2.6/100,000 indigenous people in Colombia and
3.6/100,000 indigenous people in the department of Cauca.
The ARSB
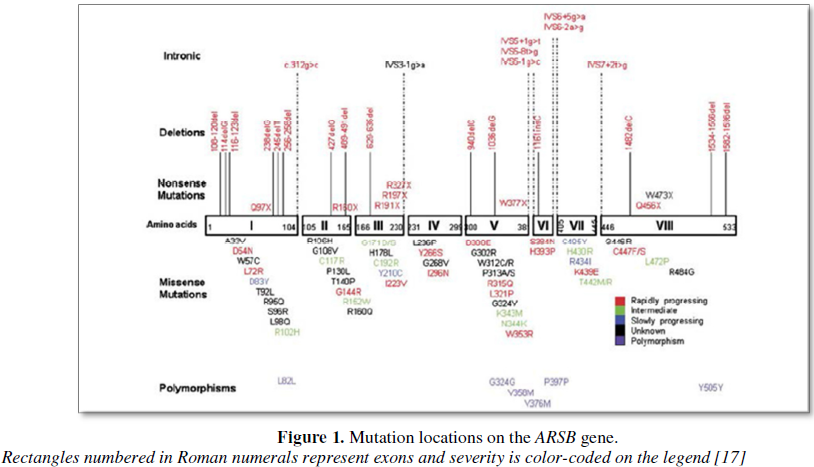
human gene is a 209 kb gene comprised of 8 exons, ranging in size from 71 to
885 bp [1]. It encodes a polypeptide of 533 amino acids [1]. MPS VI results
from several ARSB mutations,
including missense, non-sense, splice site, small deletions, small insertions,
insertion-deletion (indel) and large deletions [4].
A molecular study of 14 Colombian patients
with MPS VI [6] identified 14 mutations (80% with severe phenotype), of which
57% had not been previously reported (p.H111P, p.C121R, p.G446S, p.*534W,
p.S334I, p.H147P, c.900TNG, and c.1531_1553del) and 43% had private mutations
(p.G144R, p.W322*, p.G302R, p.C447F, p.L128del and c.1143-1GNC).
The Department of Cauca in southwestern
Colombia has a total population of 1,355,000 inhabitants. It is characterized
by a high incidence of some genetic disorders (such as muscular dystrophies, osteogenesis
imperfecta and hemoglobinopathies), probably due to the relative inbreeding and
the geographical isolation of these regions. The population of this department
belongs to three large ethnic groups: Afro-Colombians (22.19%), “mestizos” and
white (56.31%) and the Indigenous or Amerindian groups (Paeces, Guambianos and
Ingas, among others) (21.5%). These indigenous populations are some of the
largest in the country (28.7% of the total national indigenous population).
Here, we have identified a total of 16 cases out of the 36 reported in Colombia
for MPS type VI (45% of the cases recorded in this country) (Author data, not
published yet). The frequency reported may be higher, as many patients are not
diagnosed or die before a definitive diagnosis is made.
The main objective of this study is to
characterize, by molecular genetics, two patients identified in southwestern
Colombia with the severe form of MPS type VI.
MATERIALS AND
METHODS
Sample and family
data collection
All the 16 patients found in the Department
of Cauca have been characterized clinically and confirmed by means of enzymatic
activity test in leukocytes. Two of these patients, coming from the
municipalities of Totoró and Piendamó, were considered index cases for
assessment and clinical care under our study in the Pediatrics Service of San
José University Hospital in Popayan, Cauca. After obtaining the authorization
of the institutional Ethics Committee and the informed consent from the father,
mother or legal guardian of the children, a molecular characterization of the
mutation was performed on the index cases (DNA extraction, PCR amplification
and gene sequencing (ABI PRISM*3100 Genetic Analyzer) for each exon).
RESULTS
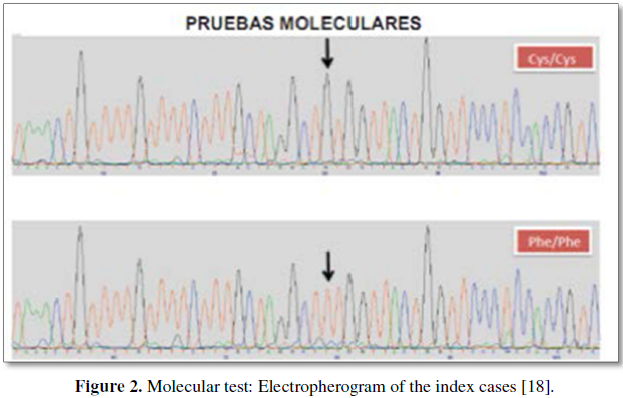
A single nucleotide (p.C447F) pathogenic
transversion producing a sense change mutation was found in the two index cases
on exon 8 of the ARSB gene, using the
ALAMUT software (version 2). This gives rise to the exchange of one amino acid
for another on the minor domain of the enzyme: position 206,029 (T/T)
TGT>TTT; mutation p.C447F (Figures 2
and 3).
DISCUSSION
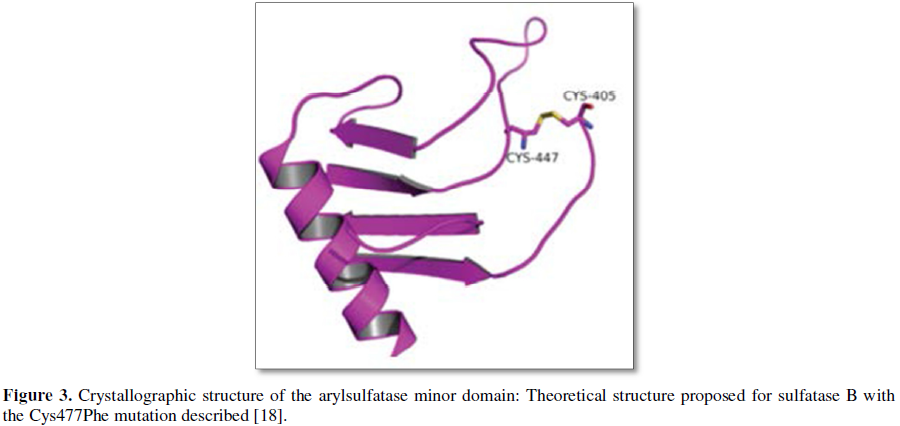
In the crystallographic structure of the
enzyme (PDB code 1FSU), the lateral chain of the C447 amino acid appears as
forming a disulphide bridge with the lateral chain of the Cys405 amino acid.
This is an important secondary structural element, and when it is lost in the
p.C477F mutation, it affects the folding ability of this domain and,
consequently, its secondary structure.
The biological function of the minor domain
appears to be related with enzyme solubility, molecular recognition of the
substrates to be hydrolyzed and bioavailability in the different organs, cells
and intracellular media. The mutation described results in a structural change
in the minor domain, with a loss of enzyme selectivity for its substrates, as
well as for changes in its solubility in the different biological media,
resulting in different bioavailability when compared to the native protein. The
three-dimensional structure of the major domain (amino acids 1-382) should not
be affected by this mutation. Nevertheless, these functions must be severely
affected by the mutation as a result of the structural change in the minor
domain. This should lead to a loss of selectivity of the enzyme for its
substrates.
An unusual frequency of genetic diseases is
found in the department of Cauca in Colombia, which is probably underestimated
due to misdiagnosis, lack of diagnosis and patients dying before diagnosis. In
this study, the two index patients exhibit the same mutation, suggesting the
possibility of a common ancestral allele, probably due to the relative
inbreeding and the geographical isolation of these regions. The above
highlights the importance of public health policies in our country, genetic
counseling, neonatal screening and identification of new cases in areas where
incidence is above average.
CONCLUSION
The two index cases from different
municipalities present the same homozygous p.C447F mutation. Given the low rate
of migration and high frequency of endogenic marriages; the finding of the same
mutation, suggests the possibility of a common ancestral allele and enhances
the importance of molecular diagnosis in the study of gene hereditary diseases,
allowing genotype-phenotype correlation which may predict severity and enzyme
replacement success.
FUNDING
Fundación Pública Galega de Medicina Xenómica
and Biomarín Colombia provided support for this study.
ACKNOWLEDGMENT
1. Litjens
T, Baker EG, Beckmann KR, Morris CP, Hopwood JJ, et al. (1989) Chromosomal
localization of ARSB, the gene for human N-acetylgalactosamine-4-sulphatase.
Hum Genet 82: 67-68.
2. Valayannopoulos
V, Nicely H, Harmatz P, Turbeville S (2010) Mucopolysaccharidosis VI. Orphanet
J Rare Dis 5: 5.
3. Lin
HY, Chuang CK, Wang CH, Chien YH, Wang YM, et al. (2016) Long-term galsulfase
enzyme replacement therapy in Taiwanese mucopolysaccharidosis VI patients: A
case series. Mol Genet Metab Rep 7: 63-69.
4. Litjens
T, Hopwood JJ (2001) Mucopolysaccharidosis type VI: Structural and clinical
implications of mutations in N-acetylgalactosamine-4-sulfatase. Hum Mutat 18:
282-295.
5. Kantaputra
PN, Kayserili H, Guven Y, Kantaputra W, Balci MC, et al. (2014) Clinical manifestations
of 17 patients affected with mucopolysaccharidosis type VI and eight novel ARSB
mutations. Am J Med Genet Part A 164: 1443-1453.
6. Giraldo
GA, Ayala-Ramírez P, Prieto JC, García-Robles R, Acosta JC (2016) Molecular
findings of Colombian patients with type VI mucopolysaccharidosis
(Maroteaux-Lamy syndrome). Meta Gene 7: 83-89.
7. Garrido
E, Chabás A, Coll MJ, Blanco M, Domínguez C, et al. (2007) Identification of
the molecular defects in Spanish and Argentinian mucopolysaccharidosis VI
(Maroteaux-Lamy syndrome) patients, including 9 novel mutations. Mol Genet
Metab 92: 122-130.
8. Saito
S, Ohno K, Sugawara K, Sakuraba H (2008) Structural and clinical implications
of amino acid substitutions in N-acetylgalactosamine-4-sulfatase: Insight into
mucopolysaccharidosis type VI. Mol Genet Metab 93: 419-425.
9. Hendriksz
CJ, Berger KI, Lampe C, Kircher SG, Orchard PJ, et al. (2016) Health-related
quality of life in mucopolysaccharidosis: Looking beyond biomedical issues.
Orphanet J Rare Dis 11.
10. Brunelli
MJ, Atallah AN, da Silva EM. (2016) Enzyme replacement therapy with galsulfase
for mucopolysaccharidosis type VI. Cochrane Database Syst Rev 3: CD009806.
11. Garrido
E, Cormand B, Hopwood JJ, Chabás A, Grinberg D,et al. (2008) Maroteaux-Lamy
syndrome: Functional characterization of pathogenic mutations and polymorphisms
in the arylsulfatase B gene. Mol Genet Metab 94: 305-312.
12. Stevenson
DA, Rudser K, Kunin-Batson A, Fung EB, Viskochil D, et al. (2014) Biomarkers of
bone remodeling in children with mucopolysaccharidosis types I, II and VI. J
Pediatr Rehabil Med 7: 159-165.
13. Giugliani
R, Harmatz P, Wraith JE. (2007) Management guidelines for mucopolysaccharidosis
VI. Pediatrics120: 405-418.
14. Jurecka
A, Zakharova E, Malinova V, Voskoboeva E, Tylki-Szymańska A. (2014) Attenuated
osteoarticular phenotype of type VI mucopolysaccharidosis: A report of four
patients and a review of the literature. Clin Rheumatol 33: 725-731.
15. Saito
S, Ohno K, Sekijima M, Suzuki T, Sakuraba H (2012) Database of the clinical
phenotypes, genotypes and mutant aryl sulfatase B structures in
mucopolysaccharidosis type VI. J Hum Genet 57: 280-282.
16. Giugliani
R, Lampe C, Guffon N, Ketteridge D, Leão-Teles E, et al. (2014) Natural history
and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy
syndrome) - 10 year follow-up of patients who previously participated in an MPS
VI survey study. Am J Med Genet Part A 164: 1953-1964.
17. Karageorgos
L, Brooks DA, Pollard A, Melville EL, Hein LK, et al. (2007) Mutational
analysis of 105 mucopolysaccharidosis type VI patients. Hum Mutat 29: 897-903.
18. Acosta
MA, Lago RM, Barros F, Carracedo AM (2016) Same mutation in two patients with
mucopolysaccharidosis type VI (Maroteaux-Lamy Syndrome) coming from different
municipalities in the Department of Cauca, South-western Colombia. J Genet
Disord Genet Rep 5: 4.
QUICK LINKS
- SUBMIT MANUSCRIPT
- RECOMMEND THE JOURNAL
-
SUBSCRIBE FOR ALERTS
RELATED JOURNALS
- Journal of Womens Health and Safety Research (ISSN:2577-1388)
- Proteomics and Bioinformatics (ISSN:2641-7561)
- Journal of Microbiology and Microbial Infections (ISSN: 2689-7660)
- Journal of Agriculture and Forest Meteorology Research (ISSN:2642-0449)
- Journal of Genomic Medicine and Pharmacogenomics (ISSN:2474-4670)
- Advances in Nanomedicine and Nanotechnology Research (ISSN: 2688-5476)
- Journal of Veterinary and Marine Sciences (ISSN: 2689-7830)


